Welcome to GenEpi’s docs!¶
GenEpi[1] is a package to uncover epistasis associated with phenotypes by a machine learning approach, developed by Yu-Chuan Chang at c4Lab of National Taiwan University and AILabs.
The architecture and modules of GenEpi.
Introduction¶
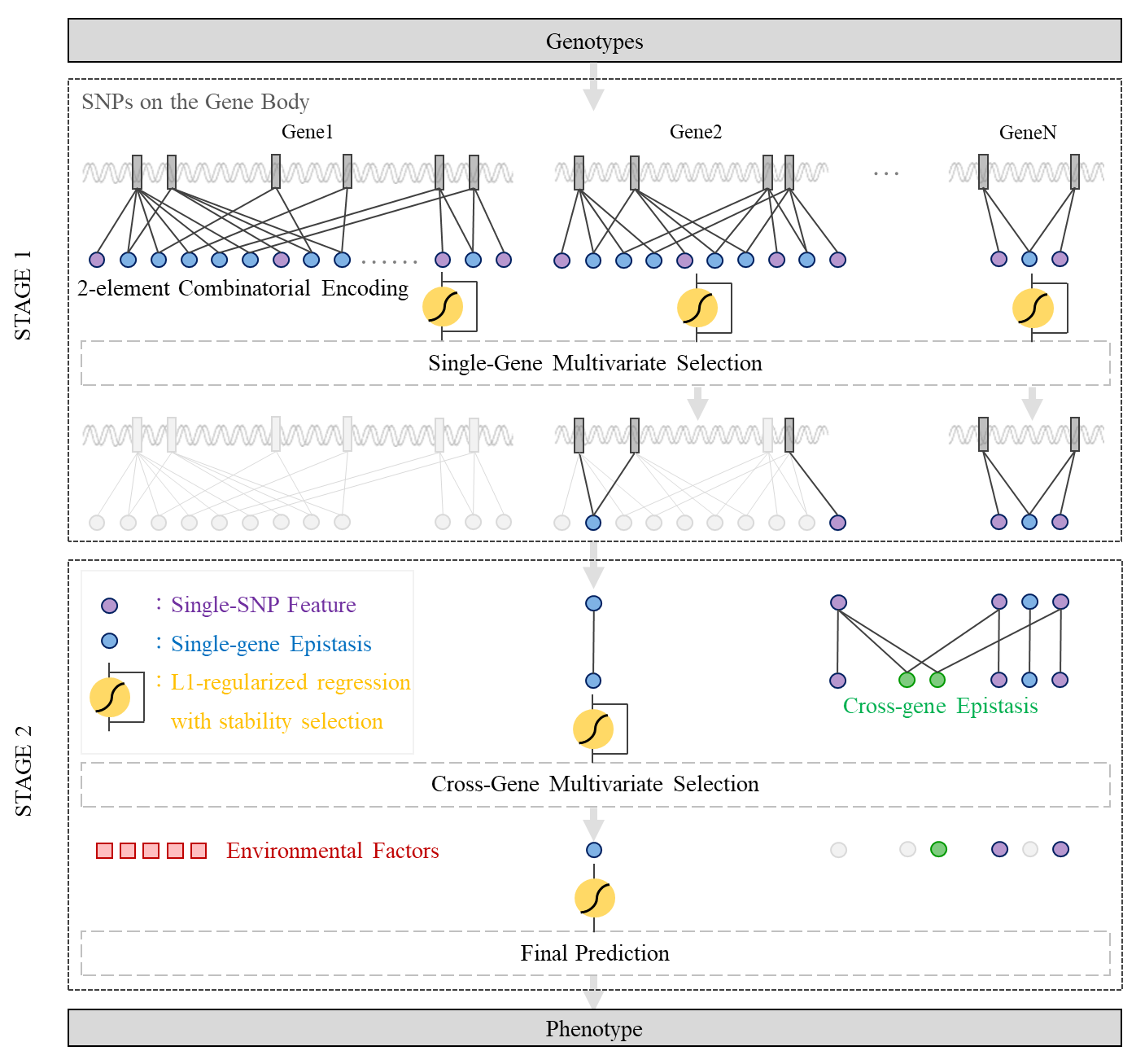
GenEpi is designed to group SNPs by a set of loci in the gnome. For examples, a locus could be a gene. In other words, we use gene boundaries to group SNPs. A locus can be generalized to any particular regions in the genome, e.g. promoters, enhancers, etc. GenEpi first considers the genetic variants within a particular region as features in the first stage, because it is believed that SNPs within a functional region might have a higher chance to interact with each other and to influence molecular functions.
GenEpi adopts two-element combinatorial encoding when producing features and models them by L1-regularized regression with stability selection In the first stage (STAGE 1) of GenEpi, the genotype features from each single gene will be combinatorically encoded and modeled independently by L1-regularized regression with stability selection. In this way, we can estimate the prediction performance of each gene and detect within-gene epistasis with a low false positive rate. In the second stage (STAGE 2), both of the individual SNP and the within-gene epistasis features selected by STAGE 1 are pooled together to generate cross-gene epistasis features, and modeled again by L1-regularized regression with stability selection as STAGE 1. Finally, the user can combine the selected genetic features with environmental factors such as clinical features to build the final prediction models.
Citing¶
Please considering cite the following paper if you use GenEpi in a scientific publication.
| [1] | Yu-Chuan Chang, June-Tai Wu, Ming-Yi Hong, Yi-An Tung, Ping-Han Hsieh, Sook Wah Yee, Kathleen M. Giacomini, Yen-Jen Oyang, and Chien-Yu Chen. “Genepi: Gene-Based Epistasis Discovery Using Machine Learning.” BMC Bioinformatics 21, 68 (2020). https://doi.org/10.1186/s12859-020-3368-2 |